
技术摘要:
本发明提供了一种基于第三代测序的单细胞转录组测序方法,所述方法包括以下步骤:(1)采用逆转录引物对单细胞RNA进行逆转录,得到带有条形码的cDNA;(2)对带有条形码的cDNA进行PCR扩增,将获得的不同单细胞来源的PCR扩增产物混合;或将不同单细胞来源的带有条形码的cDNA 全部
背景技术:
第二代测序技术(Next-generation sequencing,NGS)的出现将分子生物学研究 推向了一个高通量发展的时代,利用NGS产生了大量转录组数据,广泛应用于基础生物学研 究和医疗健康领域。传统的测序方法需要大量的起始细胞以获得足够的测序模板,得到的 数据也是所有细胞混合的结果,特别是在RNA测序中,细胞间的差异淹没在了平均值中。 单细胞RNA测序技术(scRNA-seq)应运而生,近十年来,基于NGS平台的单细胞转录 组测序取得了长足的进步,克服了研究稀有生物材料的挑战,同时揭示了生物样品的异质 性,促进了系统发育和癌症异质性等研究领域的发展。Drop-seq (Macosko EZ ,et al.Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets .Cell.2015;161(5):1202-1214;Bageritz J ,et al.Single-Cell RNA Sequencing with Drop-Seq .Methods Mol Biol .2019;1979:73–85;Zheng GX ,et al.Massively Parallel Digital Transcriptional Profiling of Single Cells.Nat Commun.2017;8:14049.)和Microwell-seq(Codina-Fauteux VA,et al.PHACTR1 Splicing Isoforms and Eqtls in Atherosclerosis-Relevant Human Cells .BMC Med Genet.2018;19(1):97.)等高度并行的scRNA-seq方法使分析人类细胞图谱(HCA)成为可 能。然而由于NGS测序平台的技术原因,读长基本不超过500个碱基,对于平均长度为1000个 碱基的信使RNA,需要进行测序数据拼接才能推断得到完整的转录本信息,难以获得不同可 变剪切的转录本信息。 第三代测序平台(TGS)克服了NGS测序平台读长短的缺点,已经应用于细胞提取总 RNA分子的直接测序。然而,TGS测序策略需要大量的原始材料来构建文库,而这些材料无法 从单个细胞中直接获得。
技术实现要素:
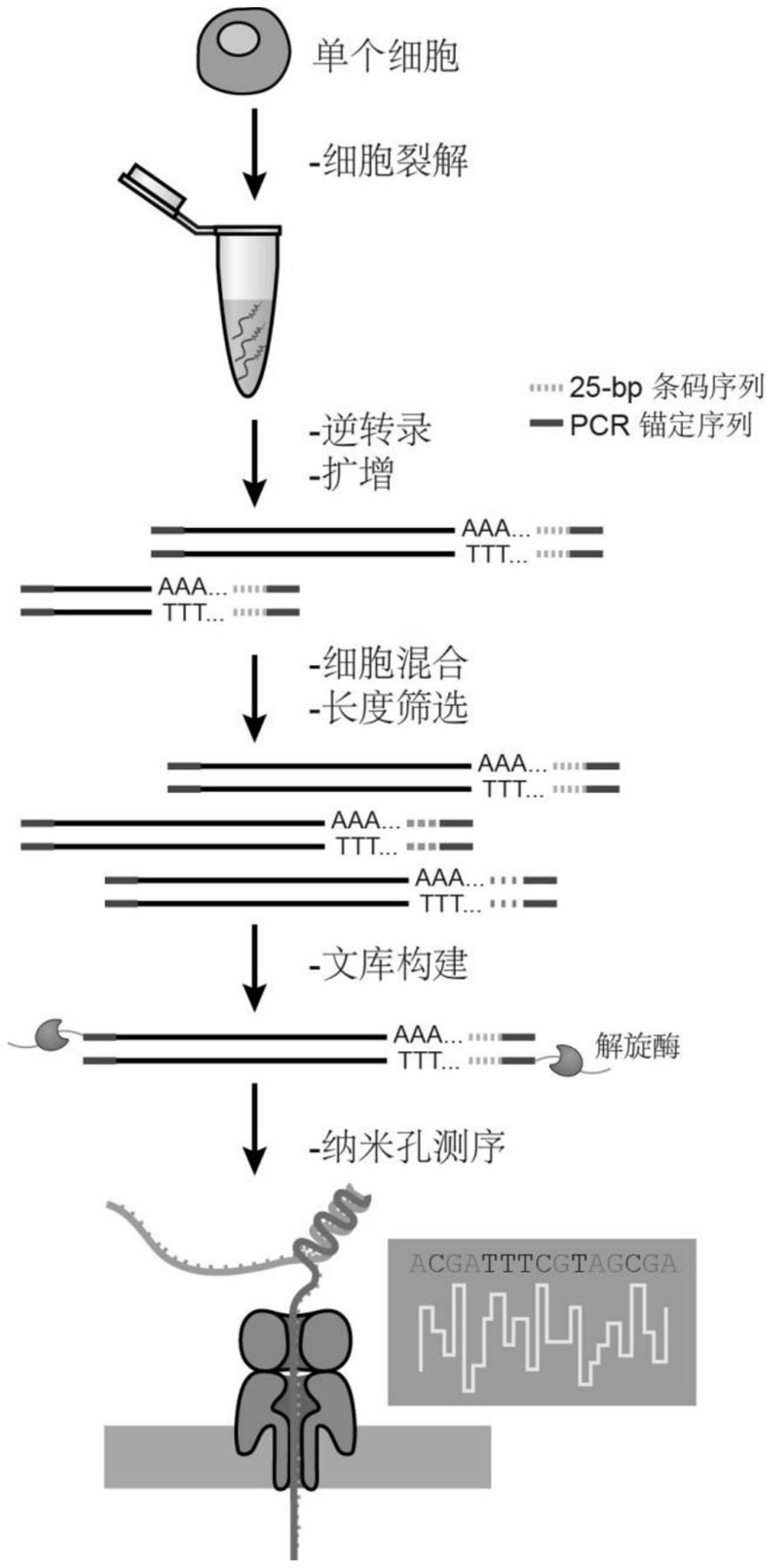
针对现有技术的不足和实际需求,本发明提供了一种基于第三代测序的单细胞转 录组测序方法,所述方法在逆转录阶段对获得的单细胞全长转录本进行条形码标记,并利 用第三代测序平台进行测序,可以实现高精度检测单细胞全长转录本的技术效果。 为达此目的,本发明采用以下技术方案: 第一方面,本发明提供了一种单细胞转录组的处理方法,所述方法包括以下步骤: (1)采用逆转录引物对单细胞RNA进行逆转录,得到带有条形码的cDNA; (2)对带有条形码的cDNA进行PCR扩增,将获得的不同单细胞来源的PCR扩增产物 混合; 或将不同单细胞来源的带有条形码的cDNA混合后进行PCR扩增。 3 CN 111549099 A 说 明 书 2/7 页 优选地,所述逆转录引物从5’端到3’端依次为锚定序列、条形码序列和polydT。 本发明中,在逆转录引物中添加条形码序列,对不同单细胞来源的RNA进行标记, 得到带有条形码的cDNA,将不同来源的产物混合后可以通过条形码序列追溯最初的单细胞 来源,利用第三代测序平台实现了对单细胞全长转录本的精确测序,解决了第三代测序平 台对原始样本需求量大的技术问题。 根据本发明,所述逆转录引物由三个部分组成:5’端的锚定序列用于向cDNA添加 相同的末端序列,有利于后续利用相同的锚定引物进行不同cDNA序列的PCR扩增;中间的与 第三代测序平台兼容的条形码序列,为不同来源的单细胞转录本添加不同的标签以进行数 据区分;3’端的多个胸腺嘧啶(T),可以有效结合到mRNA的poly(A)尾上,对转录本进行逆转 录。 优选地,所述条形码序列与第三代测序系统兼容,所述第三代测序系统例如可以 是单分子即时DNA测序、Heliscope单分子测序、基于荧光共振能量转移的即时DNA测序、纳 米孔单分子测序或离子流半导体测序中的任意一种,优选为纳米孔单分子测序。 优选地,所述条形码序列与纳米孔单分子测序系统兼容。 由于第三代测序技术的单碱基准确度略低,本发明设计了较长的条形码序列,所 述条形码序列不包含连续三个及以上的相同碱基,并且回避第三代测序偏好的错误模式, 不同条形码序列的差异足够大(至少具有10个碱基的错配);另外,采用第三代测序技术对 PCR扩增后的转录本进行测序,相当于对相同单细胞来源的转录本进行多次测序,以条形码 序列为标签进行数据分析并纠错,弥补了第三代测序技术单碱基准确度略低的问题(第三 代测序的测序错误是随机的,不像第二代测序具有测序偏向性,可以通过多次检测的数据 进行有效纠错),显著提高了测序准确性。 优选地,所述条形码序列的长度为20~40nt,例如可以是20nt、25nt、30nt、35nt或 40nt,优选为25nt。 在一种实施方式中,所述方法在将获得的不同单细胞来源的PCR扩增产物混合之 前,还包括对PCR扩增产物进行纯化的步骤。 本发明中,在混合前通过对PCR扩增产物进行纯化,去除了片段较短的产物,提高 了全长转录本的比例,利用第三代测序系统进行测序得到全长转录本的序列信息。 优选地,所述纯化采用磁珠进行。 优选地,所述磁珠与所述PCR扩增产物的体积比为(0.3~0.6):1,例如可以是0.3: 1、0.4:1、0.5:1或0.6:1,优选为(0.4~0.5):1。 优选地,所述纯化的次数为2~3次。 根据本发明,使用不同比例的磁珠进行纯化可以实现对不同片段范围的扩增产物 的筛选,本发明为筛选得到全长的扩增片段,采用的SPRI磁珠比例为样本的0.3~0.6倍体 积,纯化的次数为2~3次。 在另一种实施方式中,所述方法在将不同单细胞来源的带有条形码的cDNA混合之 前,还包括对逆转录体系进行核酸外切酶消化的步骤。 本发明中,在将不同来源的cDNA混合前,为去除逆转录体系中过量的逆转录引物, 采用核酸外切酶在35~40℃条件下消化5~15min,避免了过量的逆转录引物中的条形码序 列对结果造成的干扰。 4 CN 111549099 A 说 明 书 3/7 页 第二方面,本发明提供了一种单细胞转录组测序方法,所述方法采用第一方面所 述的方法处理单细胞转录组,并对产物进行第三代测序。 优选地,所述第三代测序包括单分子即时DNA测序(SMRT)、Heliscope单分子测序、 基于荧光共振能量转移的即时DNA测序、纳米孔单分子测序或离子流半导体测序中的任意 一种,优选为纳米孔单分子测序。 本发明中,利用逆转录引物中的条形码序列标记不同单细胞来源的cDNA,进行PCR 扩增后将不同来源的PCR扩增产物混合,或将不同来源的cDNA直接混合后进行PCR扩增,满 足了第三代测序系统对cDNA起始量的要求,实现了高精度检测单细胞的全长转录本的效 果,最后利用条形码序列有效分离不同单细胞的测序信息;同时,采用第三代测序技术对 PCR扩增后的转录本进行测序,相当于对相同单细胞来源的一个转录本进行多次测序,以条 形码序列为标签进行数据分析并纠错,弥补了第三代测序技术单碱基准确度略低的问题, 显著提高了测序准确性。 第三方面,本发明提供了一种单细胞转录组测序试剂盒,所述试剂盒包括逆转录 引物; 所述逆转录引物从5’端到3’端依次为锚定序列、条形码序列和poly dT; 所述条形码序列与第三代测序系统兼容。 优选地,所述试剂盒还包括模板转换引物、逆转录酶、dNTPs、甜菜碱或Mg2 中的任 意一种或至少两种的组合。 本发明中,所述模板转换引物从5’端到3’端依次为锚定序列和rGrG G,所述锚定 序列的核酸序列与逆转录引物中的锚定序列相同,有利于在逆转录产物的5’端和3’端添加 相同的末端锚定序列,这样在进行PCR扩增时仅需要一种引物;所述rG为核糖鸟苷, G为锁 核酸修饰鸟苷,用于增加核酸双链的稳定性并提高杂交的特异性,促进模板转换引物进行 模板转换。 根据本发明,RNA自身形成的发夹结构等二级结构可能对逆转录酶形成空间位阻, 为克服上述空间位阻,本发明在逆转录体系中添加甜菜碱,提高了逆转录酶的热稳定性和 逆转录效率。 优选地,所述试剂盒还包括锚定引物和/或PCR试剂。 根据本发明,所述锚定引物的序列与逆转录引物中的锚定序列相同。 优选地,所述PCR试剂包括DNA聚合酶、dNTPs或Mg2 中的任意一种或至少两种的组 合。 根据本发明,所述DNA聚合酶为长片段高保真DNA聚合酶,例如可以是KAPA HiFi热 启动DNA聚合酶。 第四方面,本发明提供了一种第一方面所述的方法在单细胞转录组测序中的应 用。 与现有技术相比,本发明具有如下有益效果: (1)本发明利用逆转录引物中的条形码序列,对不同单细胞来源的RNA进行标记, 将不同来源的产物混合后达到了第三代测序平台对样本起始量的要求,利用第三代测序平 台实现了对单细胞全长转录本的精确测序,获得不同可变剪切的转录本信息,而样本的最 初单细胞来源可以通过条形码序列进行确定; 5 CN 111549099 A 说 明 书 4/7 页 (2)本发明的方法可以获得单细胞中全长cDNA的准确数量,包括多种新的转录本 类型,而第二代测序方法由于需要对获得的转录本信息进行组装拼接,无法识别来自不同 转录本的组合产物,而本发明的方法可以直接读出完整的转录本序列,不同的转录本可以 被准确检测出来; (3)本发明在单细胞水平获得了不同转录本的链特异信息,对同一基因的不同转 录本可以直接进行区分,获得的测序结果敏感性高、重复性强、准确性好, (4)本发明的试剂盒和测序方法结合了单细胞全长cDNA扩增和第三代测序的优 点,在单细胞转录组分析中显示出独特的优势。 附图说明 图1为基于纳米孔单细胞测序的单细胞转录组测序流程图; 图2为对扩增产物的筛选条件进行优化前和优化后的结果图; 图3为本发明的测序方法(SCAN-seq)与二代测序(NGS-Bulk)获得的基因数量比 较; 图4为不同单细胞来源的全转录组的表达水平相关系数。











