
技术摘要:
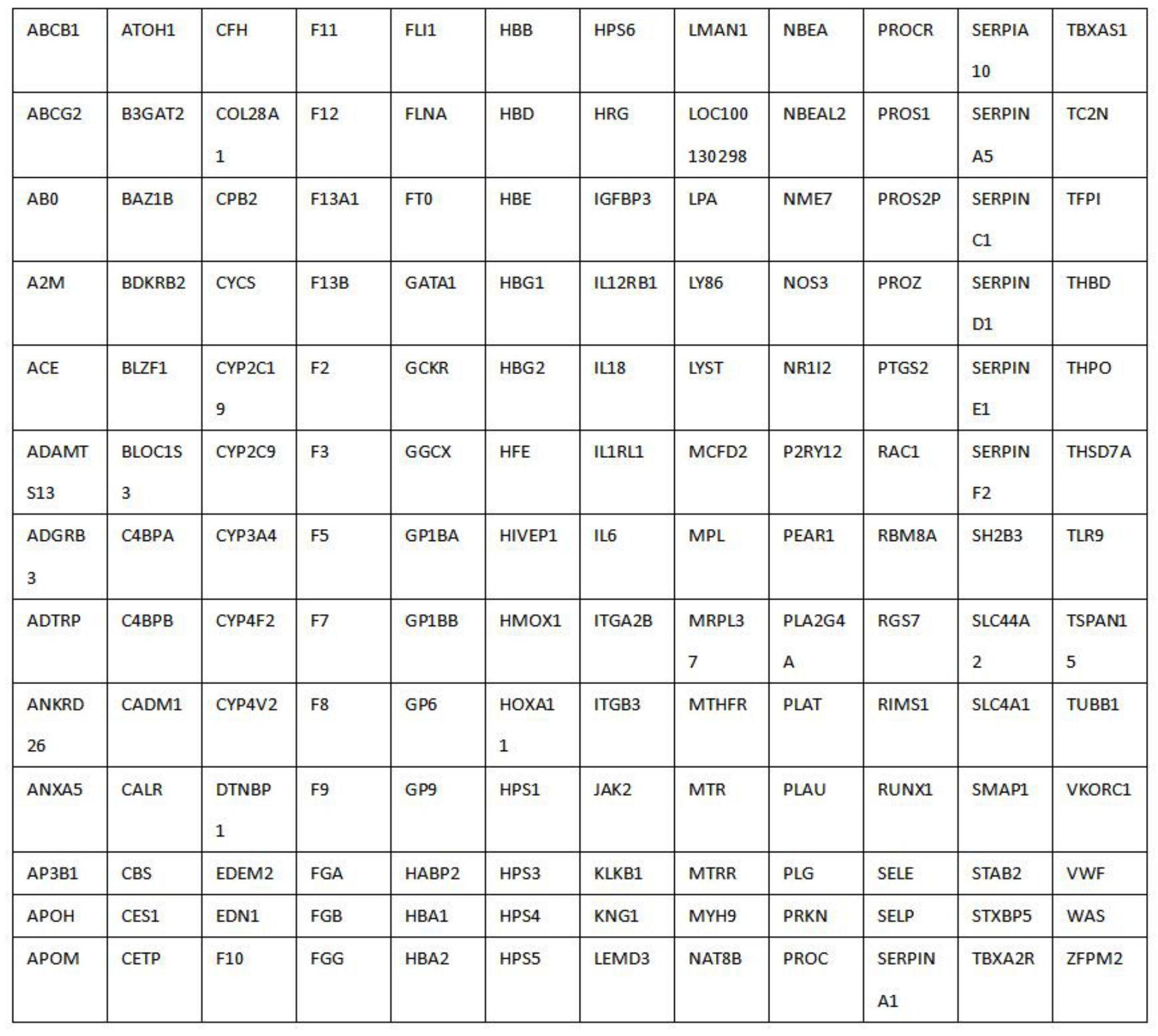
本发明公开了一种血栓与出血性疾病基因诊断方法。能够一次性全面系统地分析目前已知的156种直接或间接影响出凝血的所有相关基因,可以将重点放在人类基因组中与疾病密切相关的区域,找出致病突变。一次性针对凝血因子系统、血小板系统、纤溶系统、内皮系统、炎症系统、 全部
背景技术:
血栓与出血性疾病是由多基因环境因素引起的血液凝固状态改变,导致容易发生 血栓形成或出血的一系列疾病。血液高凝状态易导致血栓性疾病,比如:骨科和妇产科手术 后易出现的下肢深静脉血栓形成与肺动脉血栓栓塞,心内科的冠脉栓塞与急性心肌梗塞, 神经内科的脑梗塞与颅内静脉窦血栓形成。与之相反,血液低凝状态则导致出血倾向,如血 友病A、血管性血友病等。血栓形成与出血性可累及全身各器官系统,涉及临床医学多种学 科,严重危害人类健康。 尽管目前有关抗血栓药物的研究工作取得了较大进展,但当血栓形成患者就医 时,血栓往往早已形成并导致了血管栓塞,而这些脏器阻塞后再治疗效果不佳。此外血栓形 成往往由于根本病因不能得到及时明确,常影响正确的预防及治疗,从而导致严重的血栓 事件发生或复发。因此,血栓性疾病的诊疗重点在于早期诊断和基因诊断。 血栓与出血性疾病遗传倾向显著,涉及上百种血管与出凝血因子的基因异常,目 前国内外尚无针对这数百种基因进行系统性分子诊断的方法。目前最近似的相关技术也只 是针对一个或若干(20个以内)出凝血相关基因突变,设计PCR引物扩增和测序基因的启动 子区和外显子区。 目前通过多重PCR扩增建库实现对目的区域的检测,申请号201710187401 .6的专 利公开了检测ADAMTS13基因全外显子的试剂盒和方法。试剂盒包括扩增覆盖ADAMTS13基因 全外显子序列的26对正、反向引物,PCR扩增的上游引物5’端和下游引物5’端分别加了一段 长18bp的M13-F引物序列和长16bp的M13-R引物序列。基于采用Sanger测序法,利用1对通用 引物M13为测序引物,检测ADAMTS13基因全外显子序列的突变情况,应用于血栓性、血小板 减少性紫癜患者的基因诊断。但该类方法只能同时扩增10-20余种基因,引物设计困难,引 物特异性通常要求极高,反应条件苛刻,检测不稳定,而且存在扩增偏向性,拷贝数变异、插 入缺失变异检测会变得困难。 出凝血疾病的遗传异质性很大,涉及上百种血管与出凝血因子的基因异常,且血 栓与出血性疾病属于对立统一矛盾体,需同时分析每个个体的完整的出凝血基因才能准确 地科学判断其出血倾向或是血栓倾向的综合效应,只检测一个或若干个基因对出凝血疾病 的诊断远远不够。 因此,现有技术已经远远不能满足医学研究和临床应用的需求。尽管全基因组测 序、外显子测序能够从基因组范围上寻找致病突变,但该类方法因为数据量大,消耗的计算 资源多,一次性获得基因组上万个基因变异,其中绝大多数变异属于无临床意义的背景变 异,给数据分析增加了难度且周期长成本高。例如:全基因组测序数据一般高达90Gb,测序 深度30,外显子测序数据一般在10Gb,测序深度一般为50,因而假阳性率高。 4 CN 111549128 A 说 明 书 2/9 页
技术实现要素:
(一)解决的技术问题 本发明的目的在于提供一种血栓与出血性疾病基因诊断方法,以解决现有的出血 性疾病基因诊断方法检测基因突变扩展基因数量小,病因诊断确诊率低,检测不稳定,检测 困难且周期长成本高的问题。 (二)技术方案 为实现上述血栓与出血性疾病基因诊断方法解决现有的出血性疾病基因诊断方 法检测基因突变扩展基因数量小,疾病确诊率低,检测不稳定,检测困难且周期长成本高的 问题,本发明提供如下技术方案:一种血栓与出血性疾病基因诊断方法,包括以下步骤: 步骤1、根据人类参考基因组HG19,结合Ensembl、CCDS、Gencode、VEGA、SNP以及 CytoBand数据库,调取血栓与出血相关基因的所有编码序列以及编码序列两侧的20bp以内 的内含子序列; 步骤2、针对每个编码序列,由5’往3’方向,按照序列反向互补的原则,从第一个碱 基开始设计长度为120bp的探针序列,并且每两个相邻的探针序列之间存在60bp的重叠; 步骤3、在每个探针序列的5’端和3’端,分别添加TAGG TG TG T AGG CG C和 GTCAGCTAGTACGCA序列,形成2端带有同样序列的探针序列列表,并制备寡核苷酸混合物; 步骤4、通过PCR的方法,采用5’端带有生物素标记的(TTAGATAGGTGTGTAGGCGC)和 反向引物(TAAGGTGCGTACTAGCTGAC),对寡核苷酸混合物进行扩增,形成带生物素标记的血 栓与止血相关基因DNA探针文库; 步骤5、对探针文库进行上机测序,得到血栓与止血基因相关致病基因的测序数 据,将测序数据与到人类参考基因组HG19进行比对; 步骤6、统计测序数据的大小、比对率、重复率、质量值,计算血栓与止血基因相关 致病基因目标区域每个位置的测序深度;根据比对到目标区域的数据量,除以总数据量,获 得捕获效率的结果;根据目标区域每个位置的测序深度,分别统计测序深度≥1、≥4、≥10 及≥20的碱基数量,再将该碱基数量除以目标区域的总碱基数量,得到1×覆盖率、4×覆盖 率、10×覆盖率及20×覆盖率的指标信息。 优选的,所述步骤3制备寡核苷酸混合物采用寡核苷酸原位合成技术,在芯片上进 行寡核苷酸的大规模合成,用氨水将芯片上的寡核苷酸洗脱下列,溶于100微升的超纯水 中,形成寡核苷酸混合物。 优选的,所述步骤4的反应体系为: 5 CN 111549128 A 说 明 书 3/9 页 反应条件按以下20个周期循环:95℃持续1分钟,95℃持续15秒,48℃持续15秒,72 ℃持续15秒,72℃持续5分钟,4℃保温。 优选的,所述步骤5采用Illumina高通量测序仪Hiseq 4000,对探针文库进行上机 测序。 优选的,所述步骤5利用BWA MEM软件,将测序数据与到人类参考基因组HG19进行 比对。 优选的,所述步骤6采用samtools-1.9软件中的samtools stats工具统计测序数 据的大小、比对率、重复率、质量值;接着再用软件中的samtools depth工具,计算血栓与止 血基因相关致病基因目标区域每个位置的测序深度。 优选的,所述步骤1调取血栓与出血相关基因有156种。 (三)有益效果 与现有技术相比,本发明提供了一种血栓与出血性疾病基因诊断方法,具备以下 有益效果:本发明能够一次性全面系统地分析目前已知的156种直接或间接影响出凝血的 所有相关基因,可以将重点放在人类基因组中与疾病密切相关的区域,找出致病突变。与现 今流行的其它全基因组测序技术相比,在血栓相关目标区域覆盖度、有效数据量、捕获效率 数据利用率、平均测序深度、重复率等指标上本发明均优于全基因组测序,可以有效提高血 栓性疾病和出血性疾病的诊断率。 附图说明 图1为本发明的156种血栓与出血基因的所有编码序列图; 图2为本发明的应用实施例的突变的基因分布图; 图3为本发明的应用实施例中优势突变分布10种优势突变占明确分子诊断患者总 数的分布图。 6 CN 111549128 A 说 明 书 4/9 页











