
技术摘要:
本发明病毒序列整合的优化检测方法包括:从二代测序序列中筛选出病毒的参考序列,将二代测序序列采用BWA‑MEM算法对比以获得输入文件,输入文件中的序列比对结果包括完全匹配到宿主基因组上的序列的比对结果、完全匹配到病毒基因组上的序列的比对结果以及部分匹配到宿 全部
背景技术:
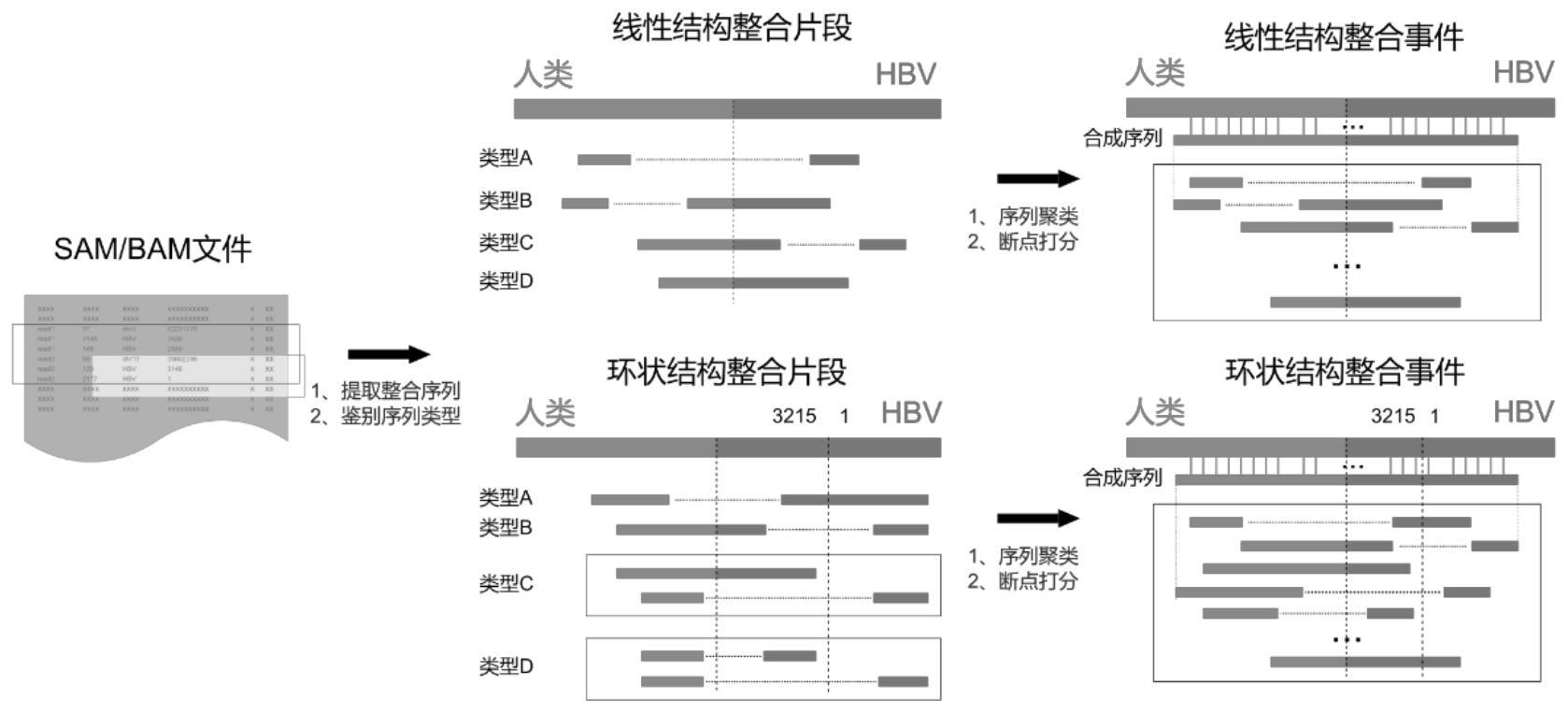
人类的癌症中,很多癌症都是与病毒感染相关的。例如,几乎所有的宫颈癌患者都 会感染人乳头瘤病毒(human papillomavirus,HPV),而在女性罹患的病毒感染相关的50% 左右的癌症中均可以检测出HPV。又例如,在全世界范围内的肝癌患者中,74%的病例感染 乙肝病毒(hepatitis B virus,HBV)或(和)丙肝病毒(hepatitis C virus,HCV)。 目前,主要存在三个问题阻碍了整合病毒整合算法与流程的开发。 1、由于病毒在复制过程中突变率很高,因此,在宿主体内会形成含有不同突变谱 系的病毒准株。在这种情况下,病毒的参考序列很难定义。早期的研究往往在数据库中下载 一条病毒序列,将其作为研究中所有样本的病毒参考序列。这种做法会在测序数据进行比 对时得到欠佳的比对结果,从而造成信息的损失。再后来的一些研究会混合多条病毒序列, 将其共同作为病毒参考序列。这种做法虽然可以避免之前研究的一些缺点,但是由于病毒 序列之间高度的相似性,会导致序列比对软件误认为序列比对至多出,而在计算比对质量 时直接输出0。这种情况的发生会导致后续的质量控制与数据处理更加复杂。针对这一现 象,VERSE流程中针对每个样本的实际数据都会得到一条相应的最优序列。如此一来,较好 的解决了上述问题。软件ViFi则是结合了隐马尔科夫链算法与传统的比对方法对病毒进行 了联合比对,从而提高比对的敏感度。虽然两种方法都较好的解决了这一问题,但是VERSE 和ViFi流程中对应的整合决策算法都没有很好的利用比对结果,从而造成流程的敏感度欠 佳。 2、比对软件的选择问题。在既往研究中,通常是选用BWA软件对测序数据进行比 对,但是BWA软件在处理一端比对至宿主(例如人)另一端比对至病毒的序列时,只会输出一 端的比对结果,序列的剩余部分以软修剪(Soft-clip)的方式输出。因此,早期的软件往往 会将这类序列提取出来,然后使用另一种甚至更多的比对软件进行第二轮比对,从而提取 相应的整合信息。但是,使用多种软件进行比对造成2个问题。首先,比对结果格式会不一 致,从而给接下来的数据处理造成困难。另外,过多使用第三方软件也会导致软件在安装过 程中难度增加。目前,随着BWA算法的更新换代,BWA-MEM是使用较多的比对软件之一。相对 于原始的BWA软件,BWA-MEM可以直接输出整合序列两端的比对结果,方便后续的数据处理。 Virus-Clip、ViFi和VICaller三个软件都使用了BWA-MEM,但是它们都没有充分的利用BWA- MEM的输出信息。 3、很多病毒的基因组都是环状的(例如:HBV和HPV)。但是,目前很少有算法专门考 虑了这一特征。通常,整合的测序序列会根据其坐标进行某种聚类,然后形成大小不同的 类,代表各个整合事件。当一个类中,病毒的序列跨越了病毒的接合点(例如:跨过了HBV的 最后一个碱基,然后从第1个碱基开始重新开始坐标计数),此时序列会主要分布于病毒的 3 CN 111584003 A 说 明 书 2/7 页 最末端与起始端。当然,也有一部分序列会直接跨过接合点。由测序数据合成的序列、整合 事件的坐标范围以及支持整合事件的序列数量,这三者对于后面的实验验证是至关重要 的。然而,在处理这类整合事件时,如果没有正确的处理会导致合成序列短、坐标范围变小 以及序列数量减小,甚至影响后面的结论。
技术实现要素:
本发明针对现有技术存在的问题和不足,提供一种新型的病毒序列整合的优化检 测方法。 本发明是通过下述技术方案来解决上述技术问题的: 本发明提供一种新型的病毒序列整合的优化检测方法,包括以下步骤: S1、首先,从二代测序序列中筛选出病毒的参考序列,并将该序列作为一条染色体 与宿主基因组序列合并,形成一个混合参考序列库,然后,采用BWA-MEM算法将二代测序序 列与混合参考序列库对比以获得SAM/BAM输入文件,SAM/BAM输入文件中的序列比对结果包 括三个部分:(1)完全匹配到宿主基因组上的序列的比对结果;(2)完全匹配到病毒基因组 上的序列的比对结果;(3)部分匹配到宿主基因组、部分匹配到病毒基因组的序列(即整合 序列)的比对结果; S2、从SAM/BAM输入文件中提取整合的序列,针对这些序列进行质量控制以过滤掉 不符合预设条件中的任一条的序列; S3、针对通过质量控制的序列,自动识别每个序列的类型; S4、在完成整合序列类型的识别后,开始进行聚类,聚类针对宿主每条染色体、每 个构象分别进行:根据宿主的整合序列坐标进行聚类,在聚类过程中坐标重复的序列会被 过滤,这一步结束会得到若干个原始的类;针对每个类,根据其中病毒序列的坐标,对每个 类进行精炼,若病毒序列距离太远,将二者分配至两个类中;根据每个类的宿主与病毒的坐 标,将距离较近的类进一步合并; S5、针对每一个类进行断点的决策,根据每个类中断点处的不同情况实施不同的 操作,将断点归为4种类型: A、准确的断点,类中的序列可给出准确的断点坐标; B、同源区域,类中的宿主序列与病毒序列重合的序列中最远序列点作为断点坐 标; C、非模板序列,类中的宿主序列与病毒序列之间插入一段不明来源的序列,将不 明来源序列的最前端的相邻点坐标和最远端的相邻点坐标作为断点坐标; D、模糊的断点,类中的所有序列都无法给出准确的断点,将序列最远端的坐标作 为断点坐标。 较佳地,预设条件为:1、比对结果中具有比对质量较低的序列,默认阈值为30,用 户可以设置;2、序列中某部分无法比对;3、序列中某部分比对至多处;4、由双端测序序列比 对结果合成的序列片段的构象异常。 较佳地,在步骤S5之后,针对同一个类,如果有证据表明多个断点可能存在,则输 出多个断点,并且会对每个断点进行打分; 假设第i个类型A的序列与第j个类型B、C或者D的序列之间针对人类的距离为dhij, 4 CN 111584003 A 说 明 书 3/7 页 针对病毒的距离为dvij,同时支持断点j的序列数量为Nj,则对断点j的打分公式为: 其中,若dhij dvij=0,则ωij=1;若dhij dvij>0, 完成打分后, 所有的断点会根据其打分进行由高到低排序,而后距离在10bp以内的断点会进行合并。 本发明的积极进步效果在于: 1、可以充分利用比对结果,找到所有的整合序列; 2、流程合理,可以最大限度的检测出整合事件; 3、可以处理跨越环状病毒的结合点的片段; 4、可以正确的处理所有情况的断点,在无法判断断点位置真实情况的时候,可以 给出预测的断点; 5、可以输出多个断点,并对每个断点排序、打分,更加符合真实情况。 附图说明 图1为本发明病毒序列整合的优化检测方法的流程图。 图2为本发明断点的4种情况示意图。 图3为本发明较佳实施例的一个跨越HBV基因组接合点的整合事件示意图。











