
技术摘要:
本发明涉及一种在保持高纯度和均匀粒径分布的前提下大规模制备托利咪酮的方法,更具体地,涉及一种通过使用四丁基溴化铵催化剂并在乙醇中重结晶而适于工业规模制备托利咪酮的方法,该方法可以在比现有技术更短的时间内制备高纯度的托利咪酮,同时保持水含量和粒径分布 全部
背景技术:
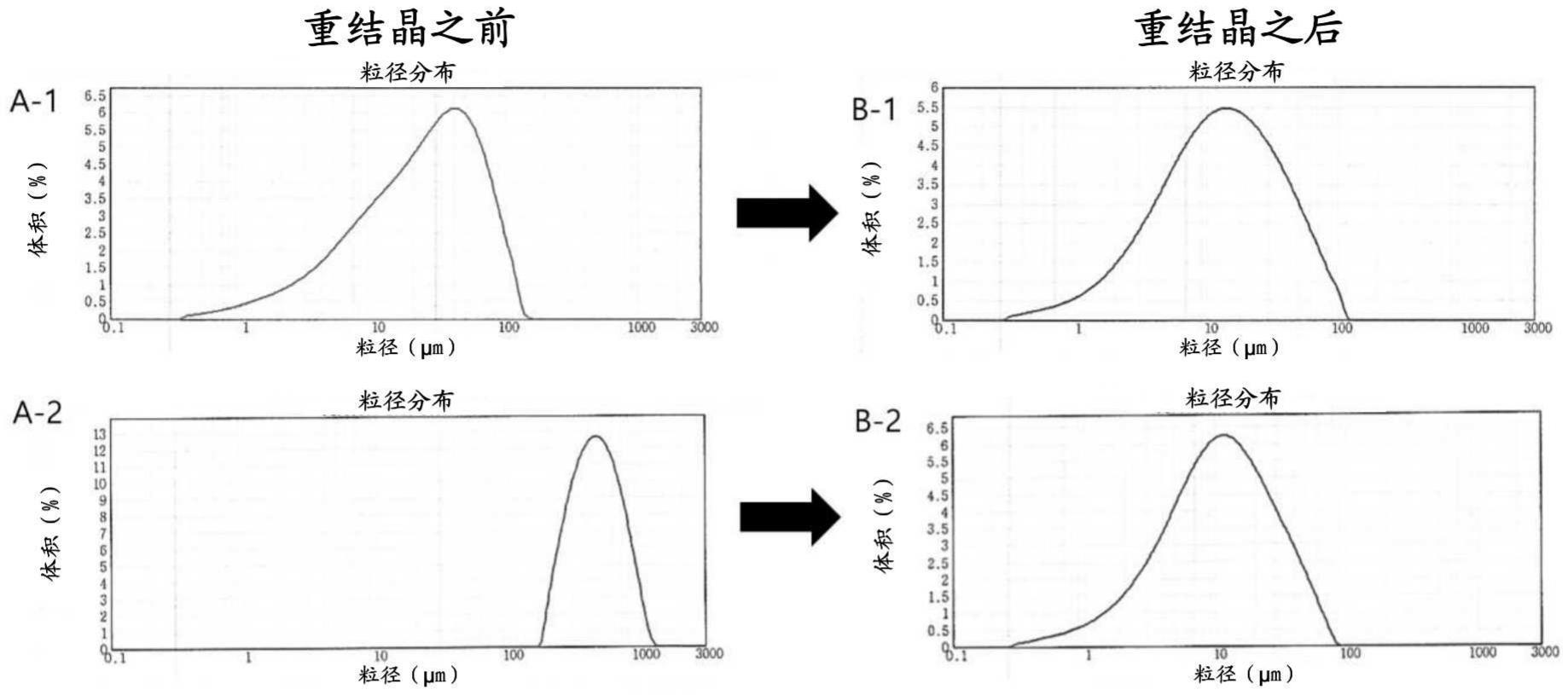
通过激活Lyn激酶通过降低血糖的作用机理,下式1的托利咪酮在糖尿病诱导的动 物模型中显示出良好的降低血糖的作用(The Journal of Pharmacology and Experiment Therapeutics,2012,Vol.342,No.1,第23-32页)。 [式1] 在目前已知的制备托利咪酮的方法中,如以下反应流程1所示,使式6和5的化合物 作为起始原料反应以获得式4的中间体,然后通过Vilsmeir反应获得式3的醛中间体。接下 来,在乙醇钠/乙醇的条件下,在搅拌下将式3的中间体和尿素回流,以获得式2的钠盐,然后 通过使用6N的盐酸或乙酸的水溶液将金属钠脱盐,从而获得式1的托利咪酮(美国专利号3, 922,345;药物化学杂志,1980,第23卷,第1026-1031页)。 [反应流程1] 然而,在上述制备方法的第一反应的情况下,为了获得式4的中间体,在迪安中存 在的情况下,应在氢氧化钾的存在下于 C的高温在Dean-Stark装置中将水从式 6的间甲酚和式5的起始原料中除去,此步骤不适用于大规模生产过程。 接下来,在通过使用盐酸或乙酸使式2的钠盐脱盐以获得式1的托利咪酮的步骤 中,通过干燥程序不容易完全除去水,该水用于大规模生产过程的过滤步骤中的洗涤。另 外,通过上述制备方法获得的式1的托利咪酮的粒径分布对于大规模生产而言不够均匀,并 且粒径分布的再现性对于大规模生产而言也不够。 众所周知,药物的粒径分布会影响药物的溶出度、生物利用度、稳定性等《( 药物科 4 CN 111587245 A 说 明 书 2/8 页 学杂志》2010,99(1), 《国际药物杂志》1995,122(1-2), )。因此,为了均 匀地维持临床使用中药物的溶出度、生物利用度等,式1的托利咪酮的均匀粒径分布非常重 要。
技术实现要素:
技术问题 本公开的目的是提供一种合适的用于大规模制备高纯度的托利咪酮,同时恒定地 保持水含量和粒径分布的方法。 本公开的另一个目的是提供一种包含通过本文公开的方法制备的托利咪酮的药 物组合物。 解决方案 为达到上述目的,本发明提供了一种制备托利咪酮的方法,包括: (i)在四取代的铵盐催化剂存在下,使式6的化合物与式5的化合物反应以制备式4 的化合物; (ii)对制备的式4化合物进行Vilsmeir反应以制备式3化合物; (iii)在搅拌下将制得的式3化合物与尿素和醇盐碱一起回流以制备式2的盐化合 物;和 (iv)使制得的式2的盐化合物脱盐以获得式1的托利咪酮,并用对应于醇盐碱的醇 将所得的托利咪酮重结晶: [式6] [式5] [式4] [式3] [式2] 5 CN 111587245 A 说 明 书 3/8 页 [式1] 其中A 是醇盐碱的阳离子。 另外,本公开提供了一种药物组合物,其包含通过上述方法制备的托利咪酮和药 学上可接受的载体。 下面更详细地解释本公开。 在根据本发明的制备托利咪酮的方法的步骤(i)中,通过在四取代的铵盐,例如四 烷基铵盐,例如四烷基卤化铵盐,例如四丁基溴化铵催化剂的存在下使式6的间甲酚与式5 的二甲基(或乙基)氯乙缩醛的起始原料反应,来制备式4的化合物。四取代铵盐催化剂反应 优选在氢氧化物碱,例如氢氧化钾的存在下,在烃溶剂,例如芳烃溶剂,如甲苯溶剂中进行。 在已知方法中,使式6的化合物和式5的化合物在Dea n - Sta rk装置中在 的高温下长时间反应,例如,当使用约160g的式6化合物和约3000g的式5化 合物时,反应进行 小时-以及从Dean-Stark装置中分离出的式4的反应物应在高温 下反馈到反应器中以使反应完全。因此,该程序不适用于大规模生产。 然而,在本公开中,当使用如上所述的相同量的式6的化合物和式5的化合物时,在 四丁基溴化铵催化剂存在下的反应可以优选在100至120℃的温度下,更优选在105至110℃ 的温度下,仅在约6小时内完成。因此,本发明适用于大规模生产。 在根据本发明的制备托利咪酮的方法的步骤(ii)中,式3的化合物通过Vilsmeir 反应由式4的化合物制备。式3的醛中间体可以通过使式4的化合物与N,N-二甲基甲酰胺和 磷酰氯反应而获得。 在根据本发明的制备托利咪酮的方法的步骤(iii)中,通过在搅拌下将式3的化合 物与尿素和醇盐碱如乙醇钠一起回流来制备式2的钠盐化合物。 在根据本发明的制备托利咪酮的方法的步骤(iv)中,将式2的托利咪酮钠盐脱盐 以获得式1的托利咪酮,并将所获得的托利咪酮在对应于步骤(iii)中的醇盐碱的醇,例如 乙醇重结晶。 在本公开中,优选通过使用盐酸或乙酸使式2的托利咪酮钠盐脱盐。脱盐之后,需 要用水洗涤的步骤,并且在洗涤之后,应该通过干燥步骤除去所使用的水。在小规模的反应 中,例如,使用15g的式3化合物的反应中,除水非常容易,使得在65℃下真空干燥14小时后 的残留水含量小于0.1%。 然而,在大规模生产托利咪酮中,例如数十至数百kg单位的产量,很难完全除去 水。在活性药物成分(API)的情形下,保持水分恒定对于控制产品质量非常重要。 在本公开中,在将托利咪酮钠盐脱盐和用水洗涤之后,没有完全干燥,将其在乙醇 中在回流下搅拌重结晶以容易地除去水。 6 CN 111587245 A 说 明 书 4/8 页 同时,通过已知方法制备的托利咪酮含有杂质,因此需要纯化过程。然而,在本公 开中,可以通过在乙醇中重结晶来去除大部分杂质。另外,在通过已知方法制备的托利咪酮 的情况下,粒径分布对于大规模生产而言不够均匀,并且粒径分布的再现性对于大规模生 产而言也是不够的,因此,当配制成药物时,可能会导致均一地保持溶出度、生物利用度等 方面的问题。然而,在本公开中,可以通过在乙醇中重结晶来使粒径分布均匀。根据本发明 制备的托利咪酮的粒径分布优选为 作为d(0.5)。 根据本公开的另一方面,提供了一种药物组合物,其包含通过上述方法制备的托 利咪酮和药学上可接受的载体。 在本公开中,药学上可接受的载体是用于肠溶目的的基质材料,并且可以是羟丙 基甲基纤维素邻苯二甲酸酯,羟丙基甲基纤维素乙酸琥珀酸酯,聚乙酸乙烯邻苯二甲酸酯, 纤维素乙酸邻苯二甲酸酯,聚(甲基丙烯酸,甲基丙烯酸甲酯)共聚物,聚(甲基丙烯酸,丙烯 酸乙酯)共聚物,虫胶或它们的混合物,但不限于上述。在药学上可接受的载体中,出于持续 释放的目的,可以使用选自疏水性材料和亲水性聚合物的组分。疏水材料是药学上可接受 的,并且可以选自聚乙酸乙烯酯,乙基纤维素和乙酸纤维素,作为聚甲基丙烯酸酯共聚物的 聚(丙烯酸乙酯,甲基丙烯酸甲酯)共聚物,聚(丙烯酸乙酯,甲基丙烯酸甲酯,甲基丙烯酸三 甲基氨基乙基酯)共聚物,脂肪酸和脂肪酸酯,脂肪酸醇,蜡等,但不限于上述。更具体地,可 以使用选自下述中的一种或多种:作为脂肪酸和脂肪酸酯的棕榈硬脂酸甘油酯,硬脂酸甘 油酯,山萮酸甘油酯,棕榈酸鲸蜡基酯,甘油的单油酸酯和硬脂酸等,作为脂肪酸醇的鲸蜡 硬脂醇,鲸蜡醇和硬脂醇等,作为蜡的巴西棕榈蜡,蜂蜡和微晶蜡等,但不限于上述。作为亲 水性聚合物,可以选择并使用糖,纤维素衍生物,树胶,聚乙烯基衍生物,聚甲基丙烯酸酯共 聚物,聚乙烯基衍生物,羧乙烯基聚合物等。具体地,作为糖可以选择和使用糊精,聚糊精, 右旋糖酐,果胶和果胶衍生物,藻酸盐,聚半乳糖醛酸,木糖胶(xylane),阿拉伯糖基木聚 糖,阿拉伯半乳聚糖,淀粉,羟丙基淀粉,直链淀粉,支链淀粉等;作为纤维素衍生物可以选 择和使用羟丙基甲基纤维素,羟丙基纤维素,羟甲基纤维素,羟乙基纤维素,甲基纤维素,羧 甲基纤维素钠,羟丙基甲基纤维素乙酸琥珀酸酯,羟乙基甲基纤维素等;作为树胶可以选择 和使用瓜尔豆胶,刺槐豆胶,黄芪胶,角叉菜胶,金合欢树胶,阿拉伯树胶,结冷胶,黄原胶 等;作为聚乙烯基衍生物可以选择和使用聚乙烯醇,聚乙烯基吡咯烷酮,聚乙烯醇缩醛二乙 基氨基乙酸酯等;作为聚甲基丙烯酸酯共聚物可以选择和使用聚(甲基丙烯酸丁酯,甲基丙 烯酸(2-二甲氨基乙基)酯,甲基丙烯酸甲酯)共聚物等;作为聚乙烯衍生物可以选择和使用 聚环氧乙烷等;作为羧乙烯基聚合物可以选择和使用卡波姆;但不限于上述。另外,如果需 要,根据本公开的药物组合物可以进一步包含例如稀释剂,粘合剂,崩解剂,流化剂,pH控制 剂等。 根据本发明的药物组合物表现出降低血糖的良好效果,因此可以有效地用于糖尿 病的预防或治疗。 发明的有益效果 本公开的制备托利咪酮的方法改进了长时间高温下的反应,以在较低温度下进行 有效减少的时间,因此非常适合大规模生产托利咪酮。根据本公开,可以在保持恒定的粒径 分布的同时制备具有低水含量的高纯度的托利咪酮。 7 CN 111587245 A 说 明 书 5/8 页 附图说明 图1显示了在乙醇中重结晶之前/之后托利咪酮的粒径分布的分析结果。 图2显示了用于测量比较例3中制备的托利咪酮的纯度的HPLC分析结果。 图3显示了用于测量在乙醇中重结晶后实施例4中制备的托利咪酮的纯度的HPLC 分析结果。 实施方式 通过以下实施例更详细地解释本公开。然而,这些实例仅出于促进对本公开的理 解而举例说明本公开,并且本公开的范围不以任何方式受到实例的限制。 实施例1-1:1-(2,2-二甲氧基乙氧基)-3-甲基苯的小规模制备(四丁基溴化铵催 化剂反应) 在1,500mL反应烧瓶中,加入间甲酚(165g,1.53mol)和甲苯(330mL),然后向其中 加入溴化四丁基铵(49.5g)。随后,缓慢加入氢氧化钾(85%,100.7g),并向其中加入氯乙醛 二甲基乙缩醛(302.1g)。将反应温度升高至110℃,并在搅拌下将混合物回流。反应6小时 后,通过TLC确认反应的终止。将反应混合物冷却至室温后,分离甲苯层,并用300mL的5%氢 氧化钠水溶液洗涤,然后用300mL的盐水洗涤。随后,将30g硫酸镁添加至有机层以除去水, 然后过滤。减压蒸发有机溶剂,得到目标化合物(278.4g,93%)。 1H-NMR 500MHz(CDCl3):7.15(m,1H) ,6.75(m,3H) ,4.68(t,1H) ,3.97(d ,2H) ,3.40 (s,6H) ,2.29(s,3H) 实施例1-2:大规模制备1-(2,2-二甲氧基乙氧基)-3-甲基苯(四丁基溴化铵催化 剂反应) 在反应器中,加入间甲酚(72.5kg),并在搅拌下加入氢氧化钾(125.43kg)。随后, 向其中加入溴化四丁基铵(21.75kg)和甲苯(145L)。加入氯乙醛二甲基乙缩醛(135.58kg) 后,将反应温度保持在105℃或以上,搅拌下回流混合物22小时(6小时后确认反应终止)。将 反应器内部冷却至 向其中加入纯净水(363L),搅拌30分钟后,将下层的水层转 移至另一反应器中。向含有水层的反应器中加入甲苯(145L),萃取后,合并有机层。向其中 加入硫酸钠(72.5kg)和硅胶(72.5kg),并搅拌1小时或更长时间。随后,在通过过滤器装置 过滤之后,将滤液转移到反应器中。在将反应器内部保持在65℃或以下的同时,将滤液减压 蒸发,得到118kg目标化合物。 比较例1:1-(2,2-二甲氧基乙氧基)-3-甲基苯的制备(使用Dean-Stark装置) 在1,000mL反应烧瓶中,加入间甲酚(160g,1.48mol),并在搅拌下缓慢加入氢氧化 钾(85%,107.4g),将混合物在 C下搅拌1小时至完全溶解氢氧化钾,然后向其 中缓慢滴加氯乙醛二甲基乙缩醛(298.8g)。在将反应混合物的温度保持在 并进行反应16小时的同时,通过使用Dean-Stark装置除去水层并将有机层加回到反应混合 物中。通过TLC确认反应终止并将反应混合物冷却至室温后,向其中加入300mL的甲苯和 400mL的纯净水。分离有机层,并用200mL的5%氢氧化钠水溶液洗涤,然后用200mL的盐水洗 涤。随后,将30g硫酸镁添加至有机层以除去水,然后过滤。减压蒸发有机溶剂,得到目标化 合物(272g,94%)。 实施例2:(E)-3-(二甲基氨基)-2-(间甲苯氧基)丙烯醛的大规模制备 8 CN 111587245 A 说 明 书 6/8 页 在反应器中,加入246.5kg的氯仿,并向其中加入131.95kg的N,N-二甲基甲酰胺。 在将反应混合物的温度保持在30℃或更低的同时,向其中缓慢地滴添加磷酰氯 (277.68kg)。滴加后,将反应混合物在55℃下搅拌2小时。向反应混合物中缓慢滴加实施例 1-2中获得的1-(2 ,2-二甲氧基乙氧基)-3-甲基苯,并在保持反应混合物温度在 的同时搅拌回流反应混合物2小时。随后,向其中加入145L甲苯,并将反应混合物 冷却至10℃或以下。向其内部保持在10℃或更低的另一反应器中,加入580L纯净水,并将反 应混合物缓慢滴加至其中。此时,内部温度保持在50℃或更低。向反应器中另外添加435L的 甲苯,并向其中缓慢滴加氢氧化钾的水溶液(836.65kg的氢氧化钾溶解在1,367L的纯净水 中)。搅拌1小时后,分离有机层,并用500L的10%盐水洗涤。将硫酸钠(72 .5kg)和硅胶 (72.5kg)添加至有机层,并将混合物搅拌1小时,然后过滤。减压蒸发滤液以除去有机溶剂, 并将136L乙酸乙酯加入到浓缩的残余物中。随后,向其中加入庚烷(29.73kg),并将混合物 在内部温度 下搅拌2小时或更长时间。将反应混合物冷却至0℃,搅拌1小时后, 将生成的固体过滤并真空干燥,得到目标化合物(80.9kg)。 1H-NMR 500MHz(Acetond-d6):8.79(s,1H) ,7.12(m,1H) ,6.92(s,1H) ,6.68-6.76 (m,3H) ,3.08(s,6H) ,2.27(s,3H) 实施例3:托利咪酮钠盐的大规模制备 在反应器中,加入(E)-3-(二甲基氨基)-2-(间甲苯氧基)丙烯醛(126千克)、尿素 (110.9千克)和乙醇(99.5%)(99.5千克),并搅拌10分钟或更长。在将反应器内部温度保持 在 的同时,向其中加入乙醇钠(21%的乙醇溶液)(688kg)。将反应器内部的温度 升高至70℃,并且在搅拌下将反应混合物回流4小时。随后,向其中加入16.4L纯净水,并将 反应混合物搅拌3小时。将反应器内部的温度降低至 将生成的固体过滤并在真 空下于65℃干燥14小时,从而获得托利咪酮钠盐(78.13kg,56.8%)。 比较例2:小规模制备托利咪酮钠盐 在反应器中,加入尿素(8.78g,0.146mol),然后向其中缓慢加入乙醇钠(21%的乙 醇溶液,54.6mL,0.146mol)。随后,向其中缓慢加入(E)-3-(二甲基氨基)-2-(间甲苯氧基) 丙烯醛(15.0g,0.073mol),并将反应混合物在约77℃下搅拌回流2小时。将2.65mL的纯净水 加入到反应混合物中,并另外在约77℃下搅拌2小时。将反应混合物缓慢冷却至室温,并将 产生的固体过滤以获得托利咪酮钠盐(9.84g,60%)。 实施例4:托利咪酮的大规模制备 向1,659L的纯净水中添加托利咪酮钠盐(78.13kg),并通过将温度升高至60℃使 其溶解,然后向其中缓慢添加乙酸(26.5kg)。将内部温度冷却至 后,将生成的固 体过滤并用630L的纯水洗涤。尽管随后在真空下在65℃下干燥32小时,但所获得的托利咪 酮(57.23Kg)的水含量为4%。随后,将干燥的托利咪酮加入到反应器中,向其中加入994L的 乙醇(99.5%),并将温度升高至70℃以使其溶解。将温度缓慢降低至 并在搅拌 下进行重结晶2小时。将反应混合物冷却至0℃,搅拌1小时,然后过滤。在65℃的烘箱中真空 干燥14小时后,得到目标化合物(44.7kg,63%)。 1H-NMR 500MHz(DMSO-d6):12.01(s,1H) ,8.30(m,2H) ,6.79-7.25(m,4H) ,2.28(s, 9 CN 111587245 A 说 明 书 7/8 页 3H) 比较例3:小规模制备托利咪酮 将在比较例2中获得的托利咪酮钠盐在60℃搅拌下溶解在150mL的纯水中。完全溶 解后,向其中滴加乙酸以在约pH 6.0下沉淀晶体。将反应混合物缓慢冷却至室温后,将沉淀 的晶体过滤并用130mL的纯水洗涤。随后,将获得的晶体在65℃的烘箱中在真空下干燥14小 时,从而获得7.6g的托利咪酮(收率:85.6%)。 比较例4:重复比较例2和3 以相同的方式重复比较例2和3,以获得7.4g的托利咪酮(收率:50.1%)。 实施例5:在乙醇中重结晶 将5.0g比较例3和4中获得的托利咪酮在搅拌回流下溶解于40mL乙醇中,缓慢冷却 至室温,搅拌2小时,并过滤以获得相应的目标化合物(4.2g,4.3g)。 实验例1:水含量测定 在65℃的烘箱中干燥小规模制备的托利咪酮(比较例3)、大规模制备的托利咪酮 (实施例4)和在乙醇中重结晶后的托利咪酮,并测定水含量。结果示于下表1。 [表1] 从上表1可以看出,在小规模制备托利咪酮的情况下,脱盐步骤中使用的水可以很 容易地被除去,但是在大规模制备的情况下,通过干燥程序不容易完全去除所使用的水。然 而,即使在大规模制备中,也可以通过在乙醇中重结晶而容易地除去水。 实验例2:粒径分析 通过使用粒径分析仪(AWM2000(MAL140253),Malvern)在以下条件下以干燥方式 测量比较例3和4中制备的托利咪酮和实施例5的在乙醇中重结晶后的托利咪酮的粒径分 布,并且结果如下表2和图1所示。 测量: -测量时间:3秒 -测量快照:3000 -背景时间:5秒 -背景快照:5000 测量周期(重复): -等分试样:每个SOP 1 -测量:每等份3 10 CN 111587245 A 说 明 书 8/8 页 -延迟:5秒 [表2] 从上面的表2和图1可以看出,在比较例3和4中制备的托利咪酮显示出不均匀的粒 径分布,并且粒径根据批次显示出很大的差异,因此该粒径分布的再现性也很差。但是,在 乙醇中重结晶后,粒径分布是均匀的。在乙醇中重结晶后,在实施例4中制备的托立咪酮的 粒径分布与B-1和B-2的相似。 实验例3:纯度测定 在以下条件下,通过HPLC分析比较例3中制备的托利咪酮和在乙醇中重结晶后的 实施例4中制备的托利咪酮,结果示于图2和3。 -色谱柱:Agilent ZORBAX Eclipse Plus C18(4.6x250mm,5μm) -检测波长:274nm -柱温:30℃ -流速:2mL/min -流动相溶剂:使用0.1%的磷酸-纯净水作为溶剂A和100%乙腈作为溶剂B的浓度 梯度条件进行分析 从图2和图3可以看出,在比较例3中制备的托利咪酮含有杂质,但是可以证实,大 多数杂质通过在乙醇中重结晶被除去。 11 CN 111587245 A 说 明 书 附 图 1/1 页 图1 图2 图3 12











