
技术摘要:
本发明涉及一种维生素B2片中有关物质的分析方法,以磷酸二氢钠溶液和甲醇与乙腈的体积比为3:7的混合溶液作为混合流动相进行梯度洗脱。所述梯度洗脱过程中流动相A和流动相B的初始比例为93~97:7~3。采用本发明的检测方法,专属性高,灵敏度高,回收率高,检出的杂质多 全部
背景技术:
维生素B2是体内黄酶类辅基的组成部分,(黄酶在生物氧化还原中发挥递氢作 用),当缺乏时可影响机体的生物氧化,使代谢发生障碍,其病变多表现为口、眼、外生殖器 部位的炎症。用于预防和治疗维生素B2缺乏症,如口角炎、唇干裂、舌炎、阴囊炎、结膜炎、脂 溢性皮炎等。 为了保证药物的安全有效,需要对药物中的有关物质进行研究、检测和监控。有关 物质主要为工艺副产物及降解产物,药品在放置过程中,杂质在发生变化,因此需要根据不 同的合成路线和生产工艺、贮藏条件建立合适的分析方法,达到对维生素B2有关物质准确、 有效的检测和监控。
技术实现要素:
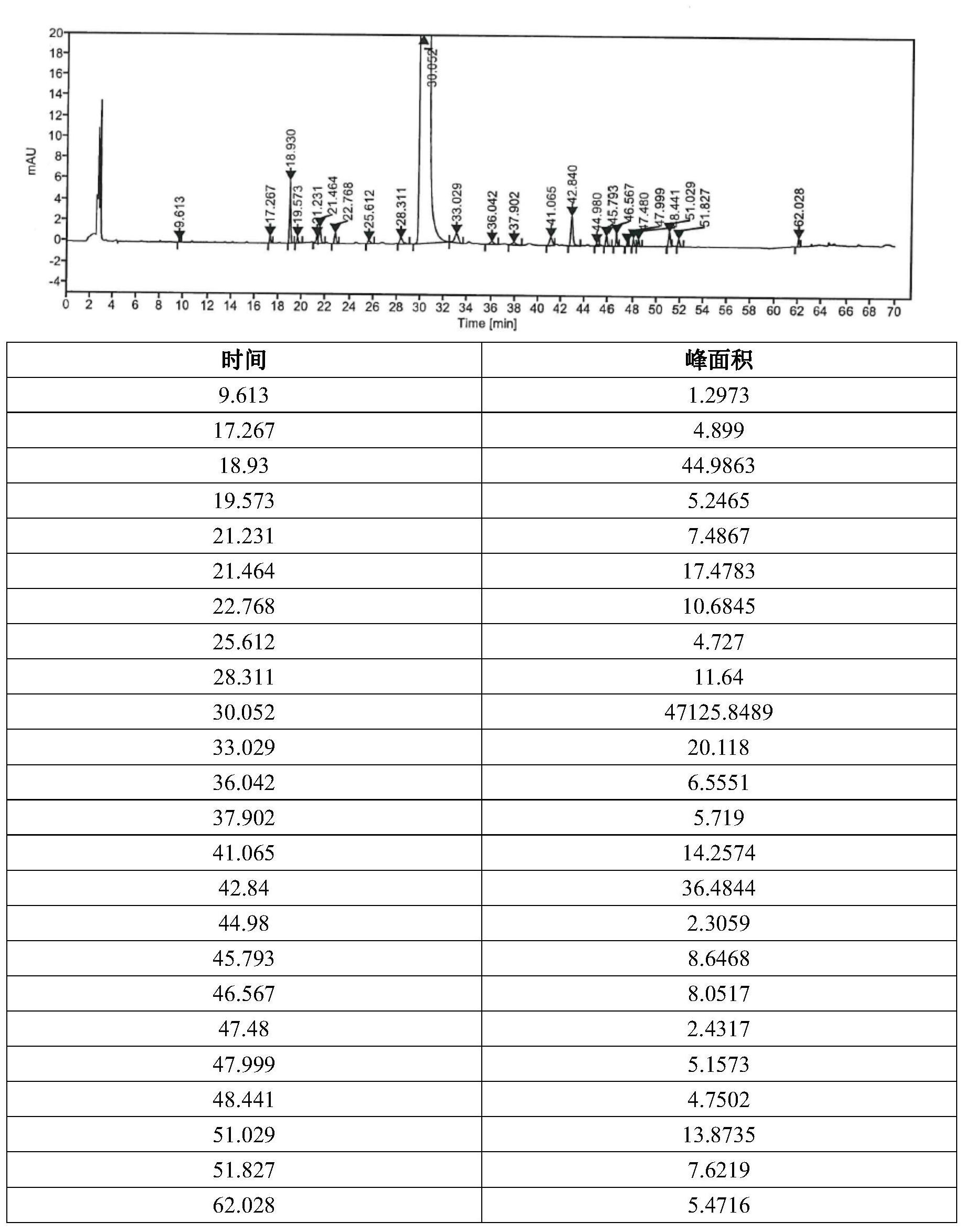
本发明的目的是在现有技术的基础上,提供一种维生素B2中有关物质的检测方 法。 本发明的技术方案如下: 一种维生素B2有关物质的检测方法,所述检测方法采用高效液相色谱对维生素B2 及有关物质进行定量检测,其高效液相色谱条件包括:采用流动相A和流动相B为混合流动 相进行梯度洗脱,所述流动相A为磷酸二氢钠溶液,所述流动相B为甲醇与乙腈的体积比为 3:7的混合溶液;所述梯度洗脱过程中流动相A和流动相B的初始比例为93~97:7~3;所述 梯度洗脱包括以下步骤:(1)在0-1分钟内,流动相A和流动相B的体积比保持初始比例不变; (2)在1-16分钟内,流动相A和流动相B的体积比由初始比例匀速渐变至85:15;(3)在16-35 分钟内,流动相A和流动相B的体积比保持85:15不变;(4)在35-45分钟内,流动相A和流动相 B的体积比由85:15匀速渐变至70:30;(5)在45-55分钟内,流动相A和流动相B的体积比保持 70:30不变;(6)在55-60分钟内,流动相A和流动相B的体积比由70:30匀速渐变至30:70;(7) 在60-65分钟内,流动相A和流动相B的体积比保持30:70不变;(8)在65-66分钟内,流动相A 和流动相B的体积比由30:70匀速渐变至初始比例;(9)在66-70分钟内,流动相A和流动相B 的体积比保持初始比例不变。 本发明的检测方法可用于检测维生素B2原料药、维生素B2片剂中的维生素B2及有 关物质。 在一种优选的方案中,当流动相A和流动相B的初始比例为95:5时,所述梯度洗脱 包括以下步骤:(1)在0-1分钟内,流动相A和流动相B的体积比保持95:5不变;(2)在1-16分 钟内,流动相A和流动相B的体积比由95:5匀速渐变至85:15;(3)在16-35分钟内,流动相A和 流动相B的体积比保持85:15不变;(4)在35-45分钟内,流动相A和流动相B的体积比由85:15 4 CN 111595961 A 说 明 书 2/19 页 匀速渐变至70:30;(5)在45-55分钟内,流动相A和流动相B的体积比保持70:30不变;(6)在 55-60分钟内,流动相A和流动相B的体积比由70:30匀速渐变至30:70;(7)在60-65分钟内, 流动相A和流动相B的体积比保持30:70不变;(8)在65-66分钟内,流动相A和流动相B的体积 比由30:70匀速渐变至95:5;(9)在66-70分钟内,流动相A和流动相B的体积比保持95:5。具 体的梯度洗脱过程如下所示: 本发明采用高效液相色谱法检测时,采用流动相A(磷酸二氢钠溶液)与流动相B (甲醇与乙腈的体积比为3:7的混合溶液)作为混合流动相进行梯度洗脱,在不影响本发明 效果的情况下,磷酸二氢钠溶液中磷酸二氢钠的浓度为0.005mol/L~0.015mol/L;优选为 0.01mol/L。 本发明中提到的流动相A的制备可包括以下步骤:将磷酸二氢钠以水溶解并稀释 成浓度为0.01mol/L,即得。 本发明中提到的流动相B的制备可包括以下步骤:将甲醇300ml与乙腈700ml混匀 即可。 本发明溶解待测样品(例如,维生素B2片剂)采用的溶剂为盐酸水溶液。例如,10% 盐酸水溶液、30%盐酸水溶液、40%盐酸水溶液、50%盐酸水溶液、60%盐酸水溶液或80% 盐酸水溶液。 本发明采用高效液相色谱法检测时,色谱柱以十八烷基硅烷键合硅胶为填充剂。 在不影响检测效果的情况下,优选色谱柱的长度为250mm,直径为4.6mm,填料粒径为5μm。例 如:YMC-PACK ODS-AQ(4.6*250mm,5μm)或Inertsustain C18(4.6*250mm,5μm)。 进一步的,高效液相色谱条件包括:检测波长为258nm~268nm,优选267nm。 进一步的,柱温为25℃~35℃,优选为30℃。 进一步的,流速为0.8~1.5ml/min,优选为1.0ml/min。 本发明可以根据需要,选择合适的进样量进样,进样量为80~100μl;优选100μl。 例如:进样量为80μl、90μl或100μl。 本发明提及的维生素B2有关物质, 采用本发明的技术方案,优势如下:包括以下物质:杂质A:7,8,10-三甲基-3,10- 二氢苯并碟啶-2,4-二酮;杂质B:7,8-二甲基-1,3-二氢苯并碟啶-2,4-二酮;杂质C:6,7-二 甲基-8-((2S,3S,4R)-2,3,4,5-四羟基戊基)蝶啶-2,4-二酮;杂质D:8-羟甲基-7-甲基-10- [(2S,3S,4R)-2,3,4,5四羟基戊基]-3,10二氢苯并碟啶-2,4,-二酮。具体如下所示: 5 CN 111595961 A 说 明 书 3/19 页 本发明采用高效液相色谱法,分别从色谱条件等方面进行筛选、优化,拟定有关物 质的检测方法,进行方法学验证,对杂质A、杂质B、杂质C和杂质D进行了定量研究,并提供完 整的验证方案,操作简单,稳定性和重现性良好。其中,杂质A的校正因子为0.7;杂质B的校 正因子为1.0;杂质C的校正因子为2.4;杂质D的校正因子为1.1。 溶解杂质A或杂质B包括如下步骤:以冰乙酸溶解杂质A或杂质B后,再以水定容及 稀释;溶解杂质C或杂质D包括如下步骤:以混合流动相溶解杂质C或杂质D后,再定容及稀 释,优选地,混合流动相中流动相A和流动相B的体积比为9:1。 本发明提供的检测方法的具体步骤为:分别配制杂质定位溶液、对照品溶液、供试 品溶液并进样,通过加校正因子的主成分自身对照法进行杂质含量检测。 本发明采用高效液相色谱法,可以配制如下样品溶液: 供试品溶液:取本品细粉适量(约相当于维生素B2片10mg)精密称定,置100ml量瓶 中,加50%盐酸水溶液5ml,振摇使维生素B2溶解,加水10ml,继续振摇数分钟,再用水稀释 至刻度,摇匀,滤过,取滤液作为供试品溶液。 对照品溶液:0.1%供试品溶液。(稀释剂:水) 6 CN 111595961 A 说 明 书 4/19 页 杂质定位溶液:称取杂质C对照品、杂质D对照品约1mg分别置10ml量瓶中,加溶剂 (流动相A:流动相B的体积比为9:1)溶解并稀释至刻度,作为杂质C与杂质D定位母液溶液; 另精密称取杂质A对照品、杂质B对照品1mg分别置100ml量瓶中,加冰乙酸(3ml)溶解后用水 稀释至刻度,摇匀,作为杂质A与杂质B定位母液溶液;取各个杂质母液加水制成每1ml中含 有对照品A0.03μg、B 0.2μg、C 0.2μg、D 0.2μg的混合溶液,作为混合定位溶液。 本发明通过筛选合适的流动相并优化流动相中各组分比例,选择合适的供试品溶 剂、杂质定位用溶剂,筛选合适的其他色谱条件,对维生素B2及有关杂质进行色谱分析,确 定了本发明的分析方法,进行了专属性(杂质与主成分分离度实验、强制降解实验)、重复 性、精密度、准确的、线性范围、校正因子、耐用性验证,确证方法的可行性。 采用本发明的技术方案,优势如下: 本发明提供的维生素B2有关物质的检测方法,克服了维生素B2在稀碱中易溶但不 稳定,而在水、乙醇、三氯甲烷或乙醚等溶剂中几乎不溶,导致的待测液难以配制的难题,在 现有技术的基础上,通过筛选合适的流动相并优化流动相中各组分比例,以盐酸水溶液作 为供试品溶剂,并优化杂质定位用溶剂,提高了待测样品的稳定性;在确保完全溶解的情况 下,以较低浓度的待测液进行色谱分析,灵敏度高、特异性强、重现性好,能够准确的分离已 知及相关未知杂质,满足维生素B2片有关物质的检测要求,可用于维生素B2片的质量控制。 附图说明 图1是杂质C以0.1mol/L氢氧化钠溶液溶解色谱图; 图2是杂质B以0.1mol/L氢氧化钠溶液溶解色谱图; 图3是杂质B以冰乙酸溶解色谱图; 图4是杂质C以流动相A:流动相B的体积比为9:1溶液溶解色谱图; 图5是空白辅料色谱图; 图6是本发明杂质混合溶液定位色谱图; 图7是本发明供试品溶液检测色谱图; 图8是EP9.0供试品溶液检测色谱图; 图9是《中国药典》供试品溶液检测色谱图; 图10是杂质A线性图; 图11是杂质B线性图; 图12是杂质C线性图; 图13是杂质D线性图; 图14是维生素B2线性图。











