
技术摘要:
本文公开了在抗微生物化合物的合成中制备硼酸酯衍生物的方法及硼酸酯衍生物的用途。本文公开的内容包括通过还原式(A)酮酯化合物的酮基来制备式(B)化合物的方法,并且该还原可以使用钌基催化剂体系或使用醇脱氢酶生物还原体系来进行。
背景技术:
技术领域 本申请涉及某些化合物以及用于制备可用于化学和医学领域中的某些化合物的 方法。更具体地,本申请涉及合成硼酸抗微生物化合物的中间体和方法。 相关领域技术 在过去半个世纪中,抗生素一直是治疗传染病的有效工具。从抗生素疗法的发展 到20世纪80年代后期,发达国家几乎完全控制了细菌感染。然而,响应于抗生素使用的压 力,多种抗药性机制已变得广泛并且威胁抗菌疗法的临床实用性。抗生素抗性菌株的增加 在大型医院和护理中心尤为普遍。抗药性菌株增加的后果包括更高的发病率和死亡率、更 长的患者住院时间以及增加的治疗费用。 各种细菌已经进化出β-内酰胺失活酶,即β-内酰胺酶,其抵消了各种β-内酰胺抗 生素的功效。根据其氨基酸序列,β-内酰胺酶可分为4类,即Ambler类A、B、C和D。A、C和D类酶 包括活性位点丝氨酸β-内酰胺酶,而较少见到的B类酶是Zn依赖性的。这些酶催化β-内酰胺 抗生素的化学降解,从而使其失去活性。一些β-内酰胺酶可以在各种细菌菌株和物种之内 和其之间转移。细菌耐药性的迅速传播和多重耐药菌株的进化严重限制了可用的β-内酰胺 治疗选项。 诸如鲍曼不动杆菌等D类β-内酰胺酶表达的细菌菌株的增加已经成为新兴的耐多 药威胁。鲍曼不动杆菌菌株表达A、C和D类β-内酰胺酶。D类β-内酰胺酶(诸如OXA家族)在破 坏碳青霉烯类β-内酰胺抗生素(例如亚胺培南)方面特别有效,亚胺培南是Merck公司 的活性碳青霉烯成分(Montefour,K.等人Crit.Care Nurse 2008,28,15;Perez, F .等人Expert Rev .Anti Infect .Ther .2008 ,6 ,269;Bou ,G .;Martinez-Beltran; J .Antimicrob .Agents Chemother .2000 ,40 ,428 .2006 ,50 ,2280;Bou ,G .等人; J.Antimicrob.Agents Chemother.2000,44,1556)。这对有效使用该类药物治疗和预防细 菌感染构成了迫切的威胁。实际上,分类的基于丝氨酸的β-内酰胺酶的数量已从1970年代 的不到十种猛增到300多种。这些问题促进头孢菌素的五“代”发展。当最初投入临床实践 时,广谱头孢菌素可以抵抗普遍的A类β-内酰胺酶TEM-1和SHV-1的水解。但是,通过在TEM-1 和SHV-1中的单个氨基酸取代的进化而产生的抗性菌株导致出现了广谱β-内酰胺酶(ESBL) 表型。 最近已经开发出了水解碳青霉烯类的抗微生物剂的新型β-内酰胺酶,包括亚胺培 南、比阿培南、多利培南、美洛培南和厄他培南以及其他β-内酰胺抗生素。这些碳青霉烯酶 属于分子A、B和D类。KPC型的A类碳青霉烯酶主要在肺炎克雷伯菌中出现,但现在在其他肠 9 CN 111556872 A 说 明 书 2/33 页 杆菌科、铜绿假单胞菌和鲍曼不动杆菌中也有报道。KPC碳青霉烯酶最早的描述见于1996年 在北卡罗来纳州,但此后在美国广泛传播。它在纽约市地区尤其成问题,据报道,有几起报 告称其在主要医院内扩散并导致患者发病。最近在法国、希腊、瑞典、英国也报道了这些酶, 而且最近在德国报道了其爆发。用碳青霉烯类药物治疗耐药菌株可能会导致相关联不良后 果。 β-内酰胺酶介导的对碳青霉烯类的抗性的另一种机理涉及结合渗透性或外排机 理与β-内酰胺酶的过量产生。一个示例是在高产ampCβ-内酰胺酶中结合的孔蛋白的损失导 致对铜绿假单胞菌对亚胺培南的抗性。外排泵过表达与ampCβ-内酰胺酶的过量生产相结合 也可能导致对碳青霉烯(诸如美罗培南)的抗性。 因此,需要改进的β-内酰胺酶抑制剂和制备这些改进的β-内酰胺酶抑制剂的有效 方法。
技术实现要素:
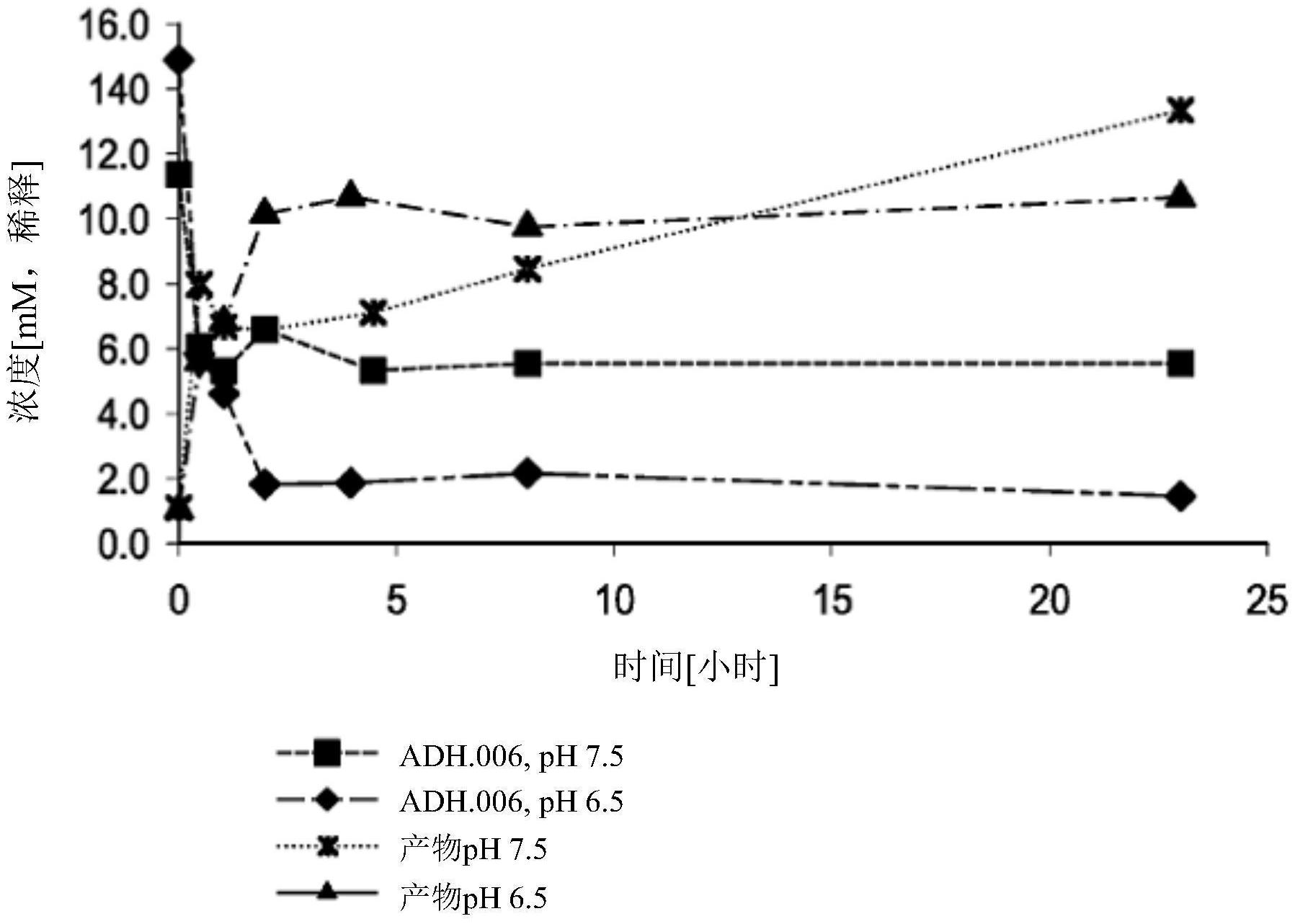
一些实施例涉及一种具有式(I)的结构的化合物: 或其盐,其中X为卤素基团,且m为2与6之间的整数。 一些实施例涉及一种具有式(II)的结构的化合物: 或其盐,其中: X为卤素基团, m为2与6之间的整数, R1a和R1b各自独立地选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取 代的C3-C8环烷基、任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取 代的杂芳基,或 R1a和R1b与居间原子一起任选地形成5-7元硼酸酯环,且 R2选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取代的C3-C8环烷基、 任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取代的杂芳基。 一些实施例涉及一种制备式(B)化合物的方法,其包括以下步骤: 还原式(A)酮酯化合物的酮基: 10 CN 111556872 A 说 明 书 3/33 页 以形成式(B)化合物: 其中: X为卤素基团,且 m为2与6之间的整数。 一些实施例涉及一种制备式(C)化合物的方法,其包括: 使硼酸酯化合物B(OR4a)(OR4b)(OR4c)与式(B-1)化合物反应 以形成所述式(C)化合物 其中: X为卤素基团, m为2与6之间的整数, R2选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取代的C3-C8环烷基、 任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取代的杂芳基,且 R4a和R4b独立地选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取代的 C3-C8环烷基、任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取代的 杂芳基,或 R4a和R4b与居间原子一起任选地形成5-8元硼酸酯环;且 R4c选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取代的C3-C8环烷基、 任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取代的杂芳基。 一些实施例涉及一种制备式(D)化合物的方法,其包括: 使镁与式(C)化合物反应: 11 CN 111556872 A 说 明 书 4/33 页 以形成第一反应中间体,以及 水解所述第一反应中间体以形成所述式(D)化合物: 其中: X为卤素基团, m为2与6之间的整数,且 R2选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取代的C3-C8环烷基、 任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取代的杂芳基,且 R4a和R4b各自独立地选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取 代的C3-C8环烷基、任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取 代的杂芳基,或 R4a和R4b与居间原子一起任选地形成5-8元硼酸酯环。 一些实施例涉及一种制备式(E)化合物的方法,其包括 还原式(A-1)酮酯化合物的酮基, 以形成式(B-1)化合物, 使硼酸酯化合物B(OR4a)(OR4b)(OR4c)与所述式(B)化合物反应以形成式(C)化合 物, 12 CN 111556872 A 说 明 书 5/33 页 使镁与所述式(C)化合物反应以形成第一反应中间体, 水解所述第一反应中间体以形成式(D)化合物, 使所述式(D)化合物与式(CL)络合剂反应 以形成所述式(E)化合物: 其中: X为卤素基团, m为2与6之间的整数, n为0与6之间的整数, Y1为O或N R9R10, Y2为O或NR11; R2选自由以下各项组成的群组:任选取代的C1-C12烷基、任选取代的C3-C8环烷基、 任选取代的C2-C12烯基、任选取代的C2-C12炔基、任选取代的芳基和任选取代的杂芳基,且 R5和R6各自独立地选自由H、任选取代的苯基和任选取代的C1-4烷基组成的群组,或 R5和R6与它们所连接的原子一起形成=O; R7和R8各自独立地选自由H、任选取代的苯基和任选取代的C1-4烷基组成的群组,或 R5和R7与它们所连接的原子一起形成芳基或杂芳基环;或R7和R8与它们所连接的原子一起 形成=O;且 R9、R10和R11各自独立地选自由H、任选取代的苯基和任选取代的C1-4烷基组成的群 13 CN 111556872 A 说 明 书 6/33 页 组。 附图简要说明 图1示出在pH值为6.5的条件下和pH值为7.5的条件下使用醇脱氢酶体系来生物还 原化合物1。 图2示出利用GDH/葡萄糖再生体系的生物还原反应过程。 图3示出利用IPA体系的生物还原反应进度。 图4示出丙酮对化合物1的还原的作用。 图5示出丙酮去除对化合物1的还原反应的作用。











