
技术摘要:
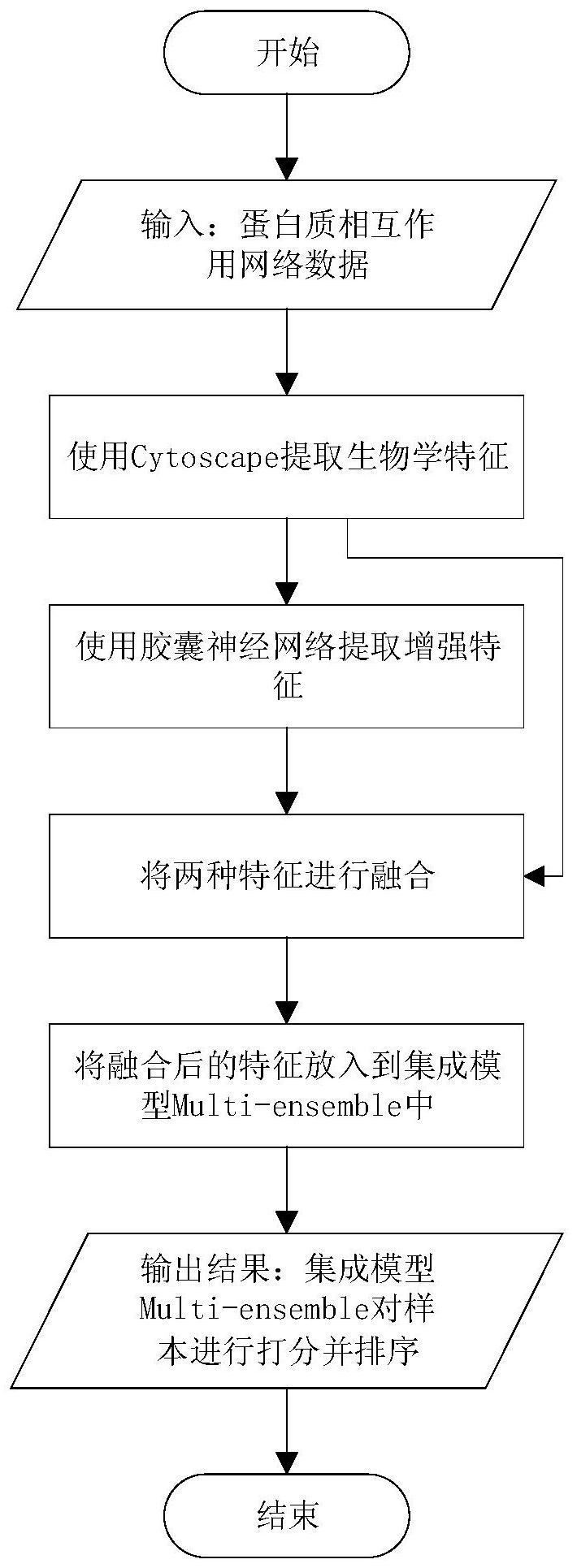
本发明公开了一种基于胶囊神经网络和集成学习的关键蛋白质识别方法,包括:步骤1:利用Cytoscape工具提取蛋白质在蛋白质相互作用网络中的八种生物学特征;步骤2:使用胶囊神经网络提取八种生物学特征的更深层的增强特征;步骤3:将生物学特征和蛋白质增强特征进行连接 全部
背景技术:
生物体的生命活动往往需要蛋白质的深度参与。关键蛋白质是指将其通过基因剔 除式突变移除后,会导致相关蛋白质复合物功能丧失,并导致细胞死亡的蛋白质。关键蛋白 质是细胞生命活动中必不可少的一部分。因此,如何准确的预测关键蛋白质成为了蛋白质 组学领域的研究重点。 早期研究关键蛋白质时,生物学家主要通过生物实验来观察生物在丢失了某些蛋 白质时对生物的影响,并以此来判断该蛋白质是否为关键蛋白质。虽然取得了不错的效果, 但是存在耗时长,耗费大等局限。为此,部分研究人员以计算机的思维来解决此类问题,再 加上高通量蛋白质组技术的快速发展和蛋白质相互作用数据的日益完善,这就使得使用计 算方法识别关键蛋白质成为了可能。Jeong等人提出了“中心性-致死性”法则,该法则将蛋 白质网络结构中的度数即相邻蛋白质节点比较多的节点称为hub点,hub点通常处于网络中 心的位置,对于整个网络的拓扑结构影响重大。而hub点的缺失对于整个网络可能是毁灭性 的,这也是在一定程度上暗示着如果hub点的缺失如同关键蛋白质缺失一样,可能会对生物 活动产生巨大的影响。基于该“中心性-致死性”法则和蛋白质相互作用数据,衍生了一批基 于蛋白质相互作用网络的中心性度量方法来度量蛋白质在网络中的特性从而识别关键蛋 白质。这些中心性包括节点的度中心性(Degree centrality,DC)。度中心性指的是网络中 某节点的领域的个数,该方法简单易用,但是预测出的关键蛋白质数量较少。节点的介数中 心性(Betweenness centrality,BC)指某节点出现在其他节点之间的最短路径的个数,它 反映了节点位置的枢纽程度,但计算的复杂度高。节点的接近中心性(Closeness centrality,CC)考察的是节点对于其他节点信息传播的依赖程度。节点的子图中心性 (Subgraph centrality,SC)利用了网络中某节点与别的节点形成的闭合回路的总数来衡 量蛋白质的节点的关键性。节点的特征向量中心性(Eigenvector centrality,EC)是利用 在网络邻接矩阵的主向量中每个顶点的分量来衡量对应蛋白质节点的关键性。节点的信息 中心性(Information centrality,IC)是利用每个顶点为端点的路径的平均总和来衡量每 个蛋白质节点的关键性。这些中心性度量方法虽然考虑了蛋白质在蛋白质相互作用网络中 的拓扑特性,但是却忽略了蛋白质本身的生物特性。 为了能更好地预测关键蛋白质,Li和Tang等人结合蛋白质相互作用网络和基因表 达信息,提出了名为PeC和WDC的关键蛋白质预测方法。Peng等人结合蛋白质相互作用网络 和蛋白质同源信息提出了ION方法。同时,又有一部分的研究采用有监督的学习方法,运用 机器学习算法,如SVM、决策树、朴素贝叶斯等来进行预测关键蛋白质。Gustafson等人通过 将具有不同预测能力的基因组特征和蛋白质特征组合,并采用朴素贝叶斯进行关键蛋白质 预测。Hwang等人基于开放式阅读框、蛋白质保守性等生物学特征和DC、BD、CC等蛋白质在蛋 3 CN 111584010 A 说 明 书 2/10 页 白质相互作用网络中的特征构建了SVM分类器来进行关键蛋白质预测。Zhong等人通过整合 蛋白质在蛋白质相互作用网络中的特征(DC、BC、CC、EC、IC、SC、NC)和结合生物特性计算出 来的特征(PeC、WDC和ION),提出了一种基于GEP的关键蛋白质预测方法。同时还有一些集成 学习算法应用到识别关键蛋白质中,Deng等[2]集成朴素贝叶斯分类器,C4.5决策树,CN2规 则和逻辑回归模型来预测关键蛋白质。Chen等[3]集成了支持向量机(SVM)和ANN来预测关 键蛋白质。Zhong等[4]融合多个XGboost分类器来预测关键蛋白质。虽然以上方法融合了一 些蛋白质的生物特性,也使用了集成学习算法来识别关键蛋白质,但是所使用的特征还是 过少,没有挖掘深层次的特征。此外集成算法只是仅仅将几个弱分类器的结果加权平均得 到最后输出。 因此需要开发更有效的特征提出方法和更有效的集成学习方法来提高关键蛋白 质的预测性能。
技术实现要素:
本发明提供了一种基于胶囊神经网络和集成学习的关键蛋白质识别方法,该方法 基于胶囊神经网络能有效提取关键蛋白质的深层次特征,并结合Multi-ensemble集成模型 能有效提高关键蛋白质识别的准确性、敏感性。 本发明的技术方案是:一种基于胶囊神经网络和集成学习的关键蛋白质识别方 法,所述方法步骤如下: 步骤1:利用Cytoscape工具提取蛋白质在蛋白质相互作用网络中的八种生物学特 征;其中,蛋白质分为非关键蛋白质和关键蛋白质两类; 步骤2:使用胶囊神经网络提取八种生物学特征的更深层的增强特征,选取胶囊神 经网络最终层获取的矩阵的第二行作为蛋白质的增强特征;其中胶囊神经网络卷积层设置 32个1×2卷积内核,步长为1,激活函数选择ReLU函数;胶囊神经网络胶囊层选取32个卷积8 维胶囊通道,胶囊神经网络从胶囊层到最终层采用了非线性激活函数执行动态路由过程; 步骤3:将步骤1得到的初始生物学特征和步骤2得到的蛋白质增强特征进行连接; 步骤4:将步骤3得到的连接后的特征放入到集成模型Multi-ensemble中,对模型 进行训练,并利用训练好的集成模型预测新的关键蛋白质; 步骤5:输出结果:将蛋白质按照由集成模型Multi-ensemble得到的打分进行降序 排序,并输出排序结果。 所述步骤2中非线性激活函数为squashing函数。 所述步骤4采用的集成模型Multi-ensemble步骤包括数据划分、挑选样本和集成 弱分类器;其中数据划分步骤将划分的训练集分为数据集P和数据集R,对数据集P进行了可 放回抽样,从而生成m个不同的数据集{P1,P2…Pm},作为m个弱分类器的初始训练集;数据集 R被划分为n个互斥子集{R1,R2…Rn},作为迭代过程的测试集,在每轮迭代过程中,如果大多 数其它弱分类器将Rj中的样本视为高质量样本,则将其添加到该弱分类器下一轮迭代的训 练集中;其中,大多数其它分类器指的是将样本视为高质量样本的其它分类器数量达到弱 分类器总个数的三分之二,j=1,2,…,n。 本发明的有益效果是:本发明使用了蛋白质在蛋白质相互作用网络中的初始生物 学特征,通过胶囊神经网络提取了这些生物学特征的增强特征,进而将初始生物学特征和 4 CN 111584010 A 说 明 书 3/10 页 增强特征结合,并在此基础上使用有效的集成方法来进行关键蛋白质的预测。本发明方法 的实验结果表明和之前基于机器学习、集成学习预测关键蛋白质的方法相比,本发明方法 提出的方法能提高识别关键蛋白质的准确性,能为生物学家进行关键蛋白质识别的实验和 进一步研究提供有价值的参考信息。胶囊神经网络提取的增强特征比初始的生物学特征更 能提高一些机器学习模型预测关键蛋白质的准确性。并且通过融合初始生物学特征和增强 特征能进一步提高机器学习模型预测关键蛋白质的准确性。 附图说明 图1为本发明方法CapsME的流程图; 图2为本发明方法CapsME中集成模型Multi-ensemble的结构图。











